
Investigating the aggregation properties of U1 small ribonucleoproteins in alzheimer's disease
Despite the known role of beta-amyloid (Aβ) in AD pathogenesis the downstream events that contribute to tau aggregation, neurotoxicity and cognitive decline still remain fundamental and poorly understood. Recently, our group reported that numerous spliceosome proteins including U1-70K aggregate in AD in close association with tau neurofibrillary tangles, but not other non-AD neurodegenerative diseases (Bai et al, PNAS, 2013). Unspliced RNA species were also found significantly elevated in AD brain, further supporting the role of spliceosome loss-of-function in AD. Aggregates of U1-70K and other core members of the U1 small ribonucleoprotein (U1 snRNP) complex are also observed in familial cases of AD caused by mutations in amyloid precursor protein (APP), which strongly links Aβ deposition with U1 snRNP aggregation (Hales et al, Mol. Neurodegeneration 2014). Given the critical role of the U1 spliceosome in regulation of 98 percent of RNA splicing events, and the large scale RNA splicing deficits in AD brain, better understanding of U1 snRNP dysfunction may reveal new signaling mechanisms linking Aβ to tau aggregation in AD. Our recent evidence indicates that intrinsically disordered low complexity (LC) domains are necessary for U1-70K aggregation in AD brain (Diner et al, JBC 2014). To better understand these mechanisms, we are currently developing new mass spectrometry approaches to map post-translational modifications (PTMs) within disordered LC domains of U1-70K and other aggregated U1 snRNPs in detergent insoluble fractions in AD brain tissue. Determining how these PTMs impact pathological U1-70K and tau associations in AD may lead to new approaches that modify global splicing defects observed in AD. These studies are being performed in collaboration with Joshua Shulman (Baylor School of Medicine), James Lah (Emory University) and Chadwick Hales (Emory University).
Discovery of novel proteomic targets for treatment of alzheimer's disease
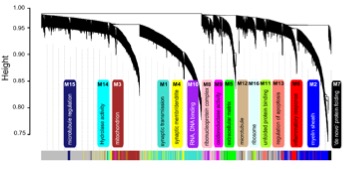
One major goal of our group is to utilize quantitative proteomics methodologies to better understand the pathogenesis of Alzheimer's Disease (AD) and other neurodegenerative diseases. As part of the National Institutes of Health (NIH) Accelerating Medicines Partnership for Alzheimer’s Disease consortium, our approach leverages the strengths of a national team of collaborating investigators and industry to couple advanced transcriptomics, proteomics and metabolomics with informatics approaches, particularly systems biology, and also employs model systems to nominate new drug targets for AD treatment. To identify novel proteomic targets, our team at Emory is processing >2,000 human postmortem brain tissues for analysis by both label-free and isobaric tagging quantitative mass spectrometry methods. Protein changes in synapses, insoluble aggregates and select post-translational modifications (PTMs), including phosphorylation and ubiquitination, will be determined. Systems biology approaches such as Weighted Co-expression Network Analysis (WGCNA) are currently being used to classify the proteome into biologically meaningful modules of co-expressed proteins linked to specific cell types, organelles, and biological pathways.
The most promising candidates will be studied for effects on neuronal viability and interactions with Aβ and tau using cell culture and drosophila models. These results and other data will drive selection of the most promising candidates to advance mouse models to assess therapeutic potential.
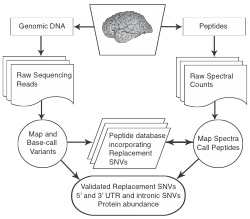
A proteogenomic approach to understanding alzheimer's disease genetic risk loci
Recent genome wide association studies (GWAS) of Alzheimer’s Disease (AD) have identified at least 20 regions of the human genome that are linked with developing AD. Currently it is not known why these regions are associated with AD. In collaboration with Thomas Wingo (Emory Neurology), we are re-sequencing genes in and around the 20 AD GWAS loci to identify all potential risk variants. Our hypothesis is that some AD GWAS signals are due to one or more single nucleotide variants (SNVs) that introduce coding changes in the translated protein product causing instability and potential aggregation. By combining our genetic sequencing data with cutting-edge mass spectrometry we will identify encoded protein products of AD GWAS candidate genes from individuals that underwent genetic sequencing. This will allow us to ask which genetic variants associate with AD and whether those genetic variants influence the abundance or aggregation potential of proteins in postmortem brain tissue of individuals with AD. This gives us the ability to directly test whether variant containing proteins are more or less abundant or aggregation prone in carriers versus non-carriers and determine whether this contributes to AD risk.