Our lab uses a broad array of biochemical, structural biology and other state-of-the-art approaches to dissect the structures and functions of biomedically important RNA molecules and their protein binding partners.
Mechanisms of bacterial antibiotic resistance
Rising antibiotic resistance among human pathogenic bacteria is a major contemporary healthcare problem. We are studying the molecular basis of how bacterial antibiotic resistance can arise through chemical modification (methylation) of the ribosomal RNA (rRNA) antibiotic binding site and through the activity of multidrug efflux systems.
Current Projects Include:
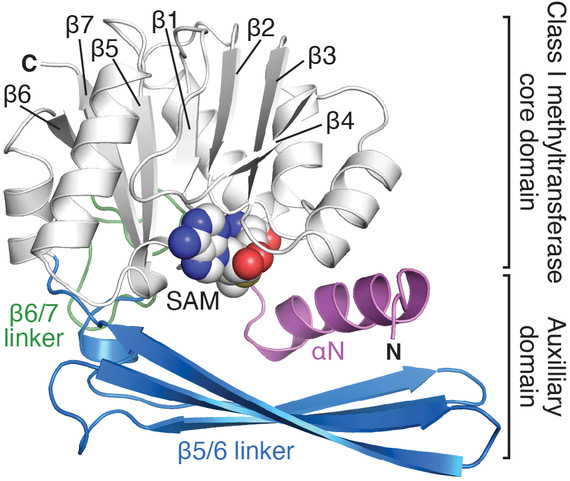
Aminoglycoside-resistance 16S rRNA methyltransferases
Aminoglycoside antibiotics typically act by binding and inducing specific conformational changes in the ribosome “decoding center” that result in aberrant protein synthesis. Methylation of the 16S rRNA drug binding site is a predominant self-protection among antibiotic-producing bacteria, and has also been increasingly identified as an acquired form of resistance in diverse human pathogens. This mechanism can confer exceptionally high-level resistance and broad specificity to a given class of antibiotics, making them a significant potential new threat. We are defining the structures of these enzymes and their complexes with their 30S substrate to understand the molecular basis for their action. The long-term goal is to use these mechanistic insights as a platform for developing new strategies to counter this emerging resistance mechanism.
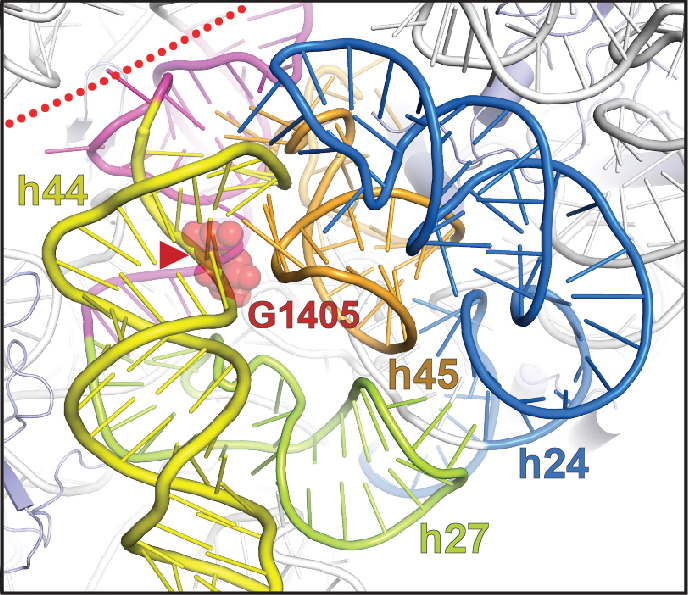
Bacterial RND multidrug efflux systems
Among several commonly known mechanisms of resistance, intrinsic multidrug efflux pumps of the Resistance-Nodulation-cell Division (RND) superfamily are broadly distributed among pathogenic bacteria (particularly Gram-negatives) and contribute extensively to clinical resistance. Efflux by the drug-inducible RND efflux pump MexXY-OprM, for example, is one of the predominant mechanisms of resistance to aminoglycoside antibiotics in P. aeruginosa and other pathogens such as Burkholderia. In collaboration with several groups at Emory (Dunham, Goldberg and Wuest) we are defining the molecular bases of MexXY gene regulation and substrate selectivity of the MexXY-OprM efflux system. Such studies may reveal novel antimicrobial targets and lay a platform for efflux pump inhibitor (EPI) development and/ or rational redesign of current treatments to evade efflux by this system.
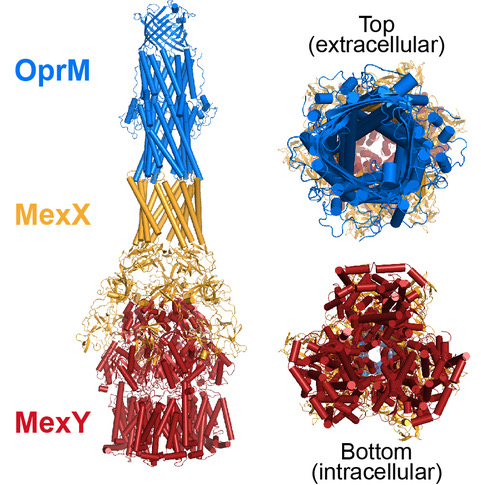
RNA-mediated regulation of human innate immune proteins
Our lab has long-standing interests in defining the structures and activities of viral and cellular non-coding RNAs and the molecular mechanisms by which they regulate proteins of the human innate immune system. In particular, our studies have centered on the double-stranded (ds)RNA sensors dsRNA-activated protein kinase (PKR) and oligoadenylate synthetase 1 (OAS1).
Current projects include:
RNA-mediated regulation of OAS
In response to dsRNA, OAS1 produces 2’-5’-linked oligoadenylate second messengers for which the only known target is the latent ribonuclease, RNase L. Activation of the OAS/RNase L pathway triggers a program of cellular and viral RNA degradation designed to halt protein synthesis in the infected cell. Our studies identified a new single-stranded RNA motif (termed 3’-ssPy) that strongly potentiates OAS1 activation by diverse RNAs, including a short model dsRNA duplex and several structured viral or cellular non-coding RNAs. Current studies in collaboration with Dr. Anice Lowen aim to fully defining the “rules” that govern potent OAS1 activation in vitro and in the context of cellular infection, using both simple dsRNAs and larger,
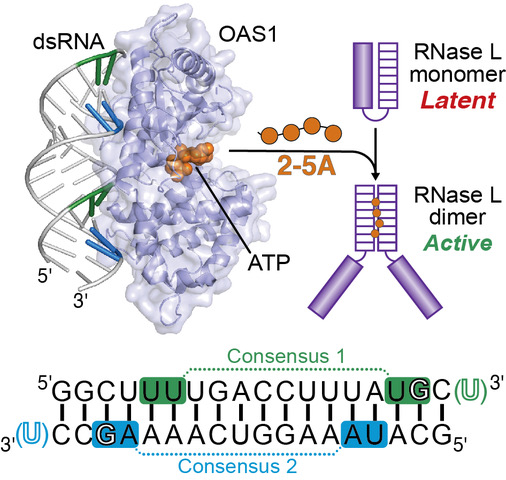
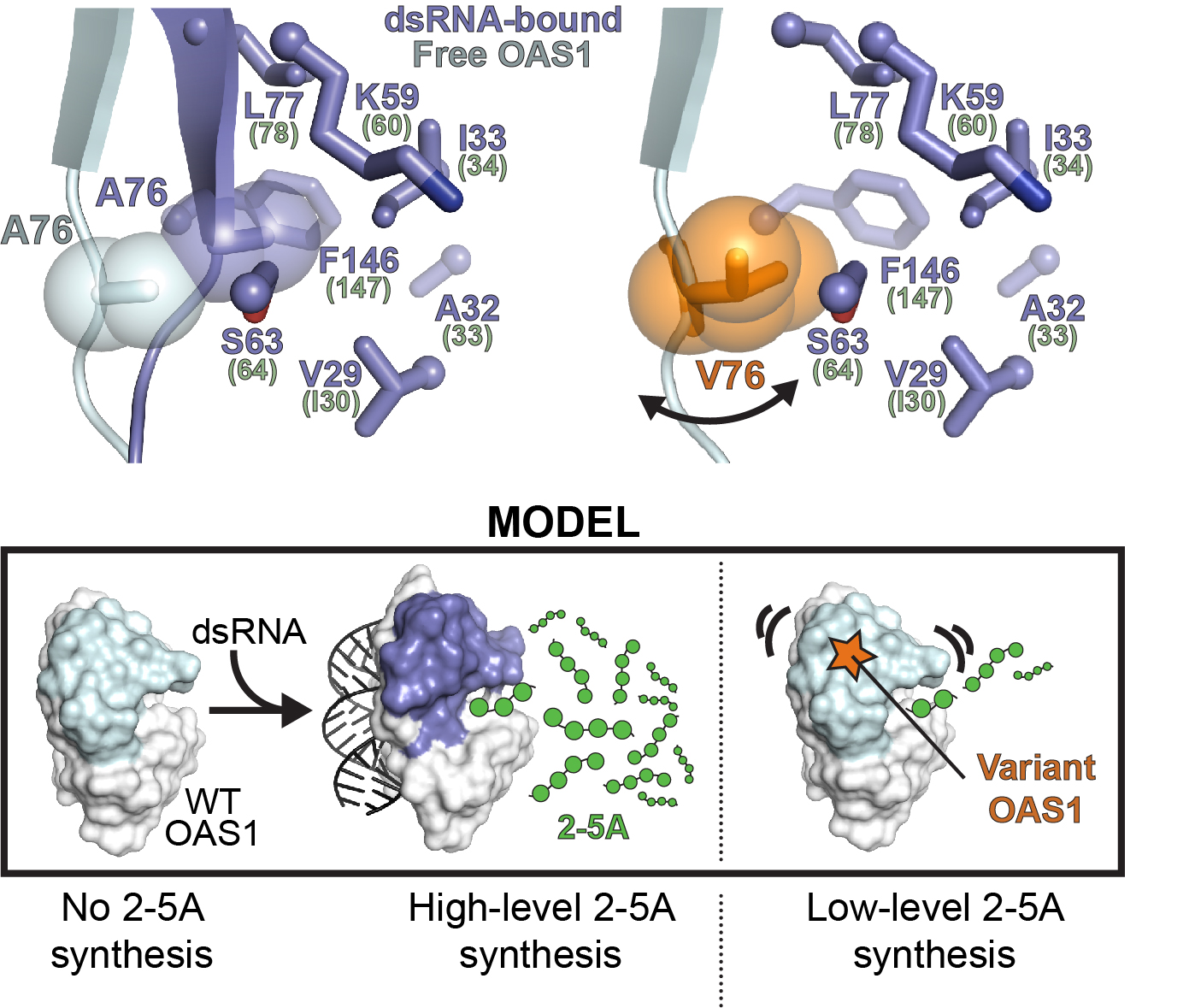
OAS1 gain of function in human disease
OAS1 activity is tightly regulated by binding of double-stranded (ds)RNA to avoid aberrant activation in the absence of viral infection or other cellular signals. We are currently working with collaborators in the US, Germany and Japan who have identified four different de novo heterozygous OAS1 variants, each of which appears to result in a common polymorphic autoinflammatory and immunodeficiency syndrome in the unrelated human patients. Our lab’s goal is to understand the molecular basis for OAS1 dysregulation resulting from these four amino acid substitutions which all result in low-level OAS1 gain-of-function activity (i.e. dsRNA-independent activity) and thus constitutively activated RNase L.
RNA and protein methylations in biology and disease
In collaboration with other labs at Emory and elsewhere, we are investigating the molecular mechanisms and biological impacts of other RNA and protein modification processes.
Current projects include:
tRNA modification by Trm10
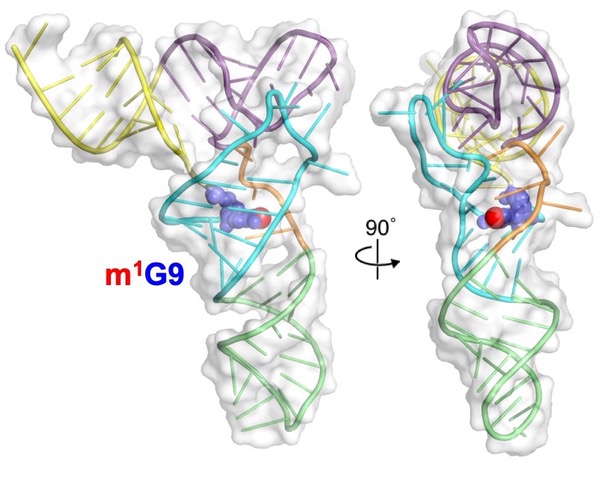
In collaboration with Drs. Jane Jackman (The Ohio State University) and Lindsay Comstock (Wake Forest University) we are investigating the structural basis for tRNA recognition and modification by yeast and human Trm10 methyltransferase. Trm10 is responsible for modifying nucleotide G9 (and sometimes A9) of many tRNAs and is conserved throughout Eukarya and Archaea. The key question we are addressing is how Trm10 specifically selects its target substrates to help generate the pool of correctly modified tRNAs.
EF-Tu Lysine 5 (K5) trimethylation in P. aeruginosa
In P. aeruginosa the translation factor EF-Tu is trimethylated on residue K5 and found on the bacterial surface, where it is proposed to mediate attachment to airway respiratory cells through platelet-activating factor receptor. In collaboration with Dr. Joanna Goldberg, whose group discovered this EF-Tu modification and the enzyme responsible (EftM), we are investigating the recognition and modification of EF-Tu by EftM, and the molecular basis of EftM temperature sensitivity which may provide a molecular “off switch” for EF-Tu modification following infection.